Working with Molecular Dynamics Panel
MD Panel of Sirius can be accessed by selecting Molecular Dynamics Panel option in the Viewers menu.
Layout and basics of trajectory visualization
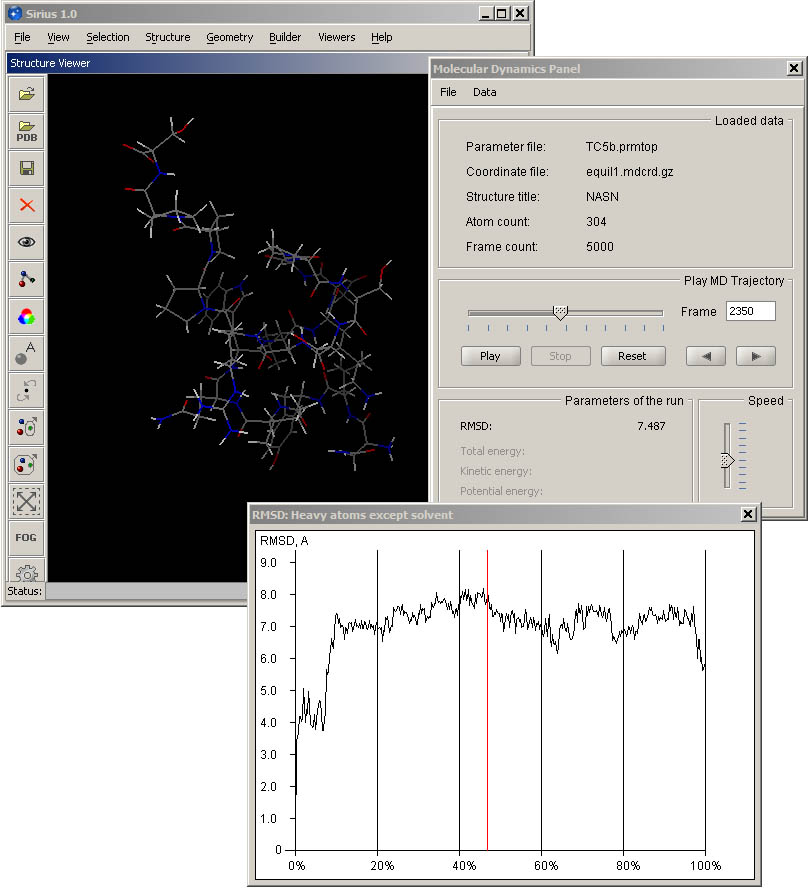
The panel consists of three main areas: loaded data, playback controls and run parameters. Sirius uses common interface for data produced by AMBER and CHARMM simulations. Once the parameter file is loaded, its name is shown in the top line of the data panel, and as soon as the coordinate file is processed and its first frame is loaded, the panel displays its name, title, atom and frame count. MD Panel can also display trajectories written as concatenated PDB file, and a specific option in the File menu is provided for this type of input.
By default, the entire trajectory is loaded, which speeds up playback and navigation between different parts of the trajectory. For example, you can jump to the desired frame by typing its number in the text field and hitting Enter, or by dragging the slider of the progress bar to the desired location. It is also possible to step through the frames one by one by using forward and backward navigation buttons under the frame counter.
If several trajectory files or one large file is being loaded, buffered loading ca be used to conserve computer memory. In this case, the application loads the first 50 frames of the trajectory and displays the number in parantheses in the Atom count field. Once playback is started, buffering will resume to maintain desired playback speed. Using buffering can be convenient when loading very large structures or long trajectories that consume considerable system memory. Note, however, that with buffered loading stepping backward is disabled. Also, you will not be able to jump forward to an arbitrary frame until the entire trajectory has been passed (buffered) at least once. Therefore, use buffering only when memory restrictions become the most important factor.
Reset returns the trajectory to its initial position. A vertical slider in the bottom right corner of the panel controls speed of the playback, and it can be adjusted at any time during playback.
Data loading
To load a trajectory, first select the Open parameter file option of the File menu. Acceptable formats include prmtop, pdb and psf. Once the parameter file is loaded, you can open the coordinate or trajectory file. The formats include inpcrd, mdcrd, restart files, and binary DCD files from CHARMM simulations. Under Data menu, a flag can be set to not load solvent if present in the input file.
Note that Sirius reads full records from binary DCD files, including any charge data. If electrostatic charge information is present, all atoms in the structure are assigned their corresponding charges, which are updated in real time as the trajectory progresses. This can be used to color structure or its parts by charge to visualize any changes.
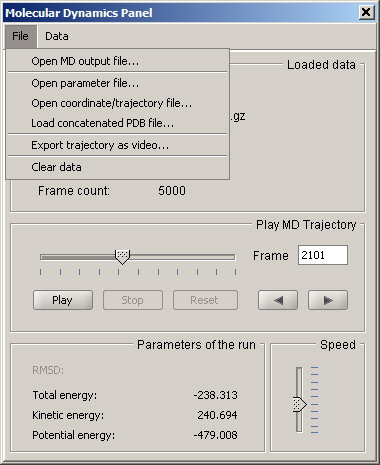
In addition to standard AMBER and CHARMM output files, Sirius can import concatenated PDB files. In this case there is no need for an additional parameter file, since the first frame serves as one.
In the case of AMBER out files that contain energy values, it is possible to display the energy graph by selecting Show energy graph in the Data menu of the MD panel.
Both types of graphs support data export: as an image or as a text file that can be selected as tab-, space- or comma-delimited (CSV).
Progress slider is used to indicate the position of the current display within the trajectory, in addition to current frame number indicator. It can also be used to fast forward or rewind to a particular location within the trajectory by dragging the slider to the desired position. Frame counter will update simultaneously to show the location. If any graphs are displayed, their markers will update accodingly. This operation can be performed any time the playback is stopped and once the entire trajectory has been played/loaded.
RMSD calculation
RMSD can be calculated for each frame in the loaded trajectory and plotted in a graph. As the default, the first frame of the trajectory is taken as the reference for calculation. Alternatively, another structure of the same molecule can be loaded by browsing the local filesystem.
It is possible to select what part of the structure is used to compute RMSD. By default, it's all loaded atoms. In addition, you can select the following sets: all atoms except solvent, all heavy atoms except solvent, protein/DNA backbone atoms, alpha-carbons of protein backbone, and custom set. In the latter case, you can use an atom set defined within the Sirius workspace (use Define/edit atom set under Structure menu). This way, you can define any set of atoms within the structure and track RMSD changes only within this substructure. For specifics on defining an atoms set refer to the corresponding help section.
While RMSD is being calculated, a message "Calculating RMSD..." is displayed in the status line of the main window. Once the operation is completed, a graph is displayed (or existing one is updated).
Exporting video and ray-traced graphics
Entire loaded trajectory or selected frames can be exported as video in QuickTime format. To export trajectory, select Export trajectory as video... in the File menu. In the dialog box, you'll be able to specify whether the entire trajectory or only a subset should be exported, as well as specify the desired framerate (number of MD frames per second of video) and name of the resulting .mov file. Trajectory export may take anywhere from several seconds to minutes, depending on the length of the trajectory, complexity of the scene and graphics hardware of your system.
Individual frames can be exported as regular images in PNG or JPG formats. We recommend PNG, since it's a lossless format that does not introduce any compression artefacts and produces relatively small files. The details of graphics export are the same as for any other loaded structure and are explained in detail in the corresponding section of help system.