Structure operations
Structure Menu Options
 |
This menu contains operations that manipulate structure objects and assist in measuring their geometry or detection of interactions. Atom set operations create and modify combinations of atoms in the user-defined context. For instance, the user may wish to define a separate set that contains a ligand and surrounding residues of the protein. Also, amino acid replacement function acts as an in silico mutation tool, which allows the user to observe the effect of the side chain replacement on the protein environment. Computation of molecular weight may be useful for quick estimates of the size of small molecules or ligands being designed for protein binding. |
Define/edit atom set
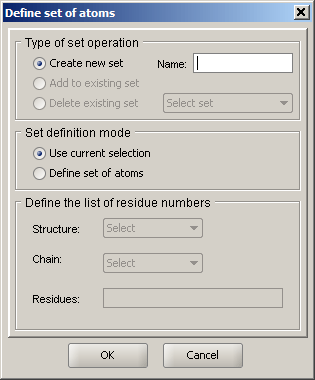 |
Sirius has a concept of a set, which is a group of atoms that may span one or more structures and has a unique name. A set, once created, can be modified to contain more atoms or deleted. Sets appear in pulldown menus of dialogs that provide structure selection, such as rendering, coloring or hydrogen bond detection dialogs. This way, a change can be applied to the predefined set, rather than to the entire structure or chain, etc. If a new set is created, a unique name must be provided. If you choose "Add to existing set" or "Delete existing set", the pulldown menu with currently defined sets will be activated. If no sets are defined, these options are grayed out. Set definition mode specifies the way to define the set: either using the currently selected part of the display (the most convenient in the majority of cases, since it offers maximum flexibility) or by selecitng structure, its chain and providing a list of atoms (similar to the mechanism used in selection by list dialog. |
Edit Structure Objects
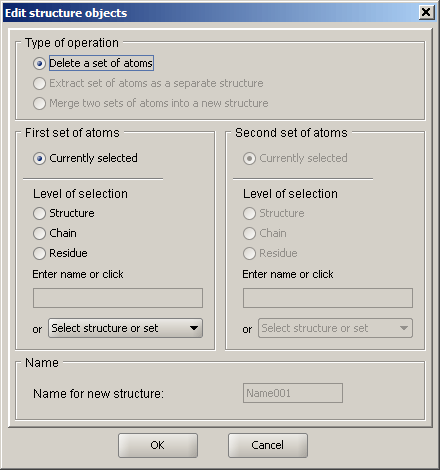
The purpose of this dialog is to provide the user with a high level control over structures and their combinations. It is intended to have options for deleting sets of atoms, combining structures into joint entities and extracting substructures. As of this release, the delete function has been implemented.
Selection of the set to which the action applies is very similar to that used in the appearance dialogs, except that it does not include individual atoms. At this point, a set of atoms that the user would like to remove from the structure can be defined and deleted. If the structure is saved to a coordinate file, only the remaining atoms will be saved.
Replace Amino Acid
In order to replace an amino acid in a peptide chain, the target residue should be clicked in the structure display and the new amino acid selected in the residue palette. The new side chain is added in a default orientation, therefore a manual adjustment may be necessary to avoid steric clashes.
Structure alignment
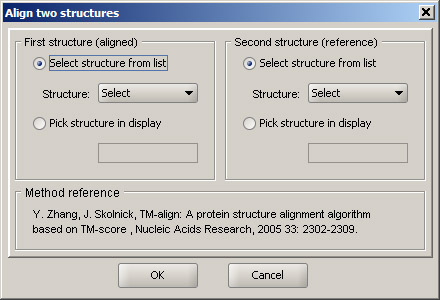
Sirius can align protein structure using a bundled implementation of TM-align developed by Y. Zhang and J. Skolnick. A reference to the original publication is provided.
In order to run a pairwise structure alignment, two structure need to be specified. The first structure is the one being moved - it's coordinates will be changed. The second structure is the reference, and coordinates of its atoms remain intact. In effect, the first structure is aligned to the second one, and the 3D display, as well as the sequence view are updated accordingly, once the alignment is completed. Frequently, result of a structure alignment is better visible when a ribbon is displayed.
Ramachandran plot
When a structure of a protein/peptide is present, it is possible to generate a Ramachandran plot that is displayed in a separate window. The Ramachandran plot viewer is interactively coupled with the other components in the Sirius environment, therefore any residue selection or coloring event causes the corresponding change to residue markers on the plot. Likewise, selecting a rsidue marker in the plot will select the corresponding residue in the structure display and sequence viewer.

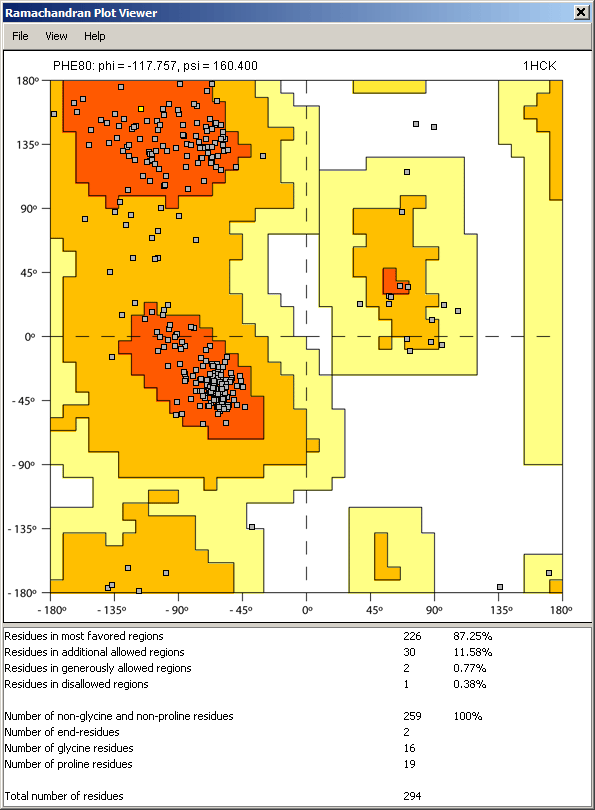
Dynamic plot update: When one of the angles in the residues is being changed by bond rotation, the plot is updated in real time to show the current location of the residue marker.
Along with the plot, statistics of the computation is displayed: number of residues located in the favorable or restricted areas of the plot, as well as count of glycine and proline residues.
Ramachandran plot viewer has its own menu, which allows for control over appearance of the markers, selection of particular residue types, and (coming soon) printing and graphics export capability.
Structure superposition
Sirius has the ability to superimpose structures based on the user-supplied anchor points. This is not a structure alignment feature, but a quick superposition routine that allows for fast structure overlays. If further modification of the structure positions is necessary, one of the structures can be moved independently to adjust its location relative to the other.
The most effective way to produce a biologically meaningfule superposition of two protein structures is to run their sequence alignment using the Align feature of the sequence viewer. Once the alignment is completed, open the superposition dialog and choose from one of the two ways to define anchor points. You can manually pick the pairs of atoms that should be superimposed. However, a more effective way of doing it with protein structures is by clicking columns in the alignment that correspond to the residues that should be aligned in the 3D structure as well. The program will then overlay the two structures, so that the atoms in each pair are as close as possible.
Once the overlay is completed, it may be a good idea to reset the view, since the structures will no longer occupy the entire available screen space.