Command line interface
Sirius features a command line interface that can be used in parallel with the menu-driven operations. Its primary purpose is to speed up the most common visualization tasks and enable saving sequences of commands to produce desired scenes that are compatible with other applications (such as RasMol).
The command syntax is based on the set of keywords used by RasMol, with the additional commands that are used to operate multiple loaded structures, directly specify desired rendering style and quality, as well as add atoms to an existing selection instead of replacing it.
Important note: unlike RasMol, SIrius uses coordinate axis direction and rotation conventions normally accepted in computer graphics and molecular visualization software. For instance, Y axis in Sirius is pointing upwards, while in RasMol it's directed down. Additional differences concern direction of rotation. Therefore, commands for translation along the Y axis, as well as rotation around the Y axis will produce in Sirius the opposite result. However, for backward compatibility, any imported RasMol scripts are automatically converted to the Sirius coordinate system, so that you will still get the same view as in RasMol. A checkbox in the Script menu indicates whether the loaded script is treated as RasMol script or not. If you start editing it and replace original commands with those specific to Sirius, be sure to uncheck the box.
Full list of Sirius commands is given in the table below. For additional description of RasMol commands please refer to the RasMol manual, since it may help in becoming aquainted with the commands used by Sirius. Also, this command line interface and script interpreter are under development, and additional commands are likely to be introduced. Any changes, however, should not affect syntax of the existing commands, which will keep any scripts compatible with the future versions of Sirius.
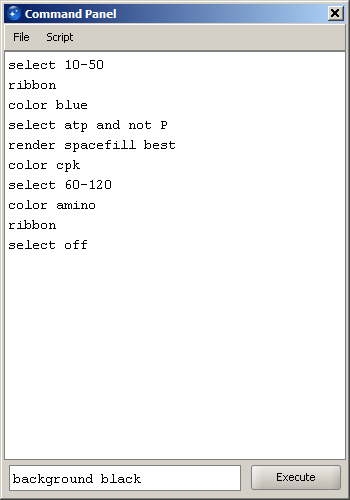 |
Command panel is activated from the Tools menu. Note that the command panel is not a separate window, and hence cannot be directly minimized. However, if it is closed or the mark in the menu is unchecked, it becomes hidden rather than deleted. Therefore, this approach can be used to temporarily hide the panel - its contents will not be lost. The panel has two parts: the large text area and the command line field under it. The command line is used to type individual commands in interactive mode. The command is run when the Execute button is clicked or Enter key is pressed. Once the command has run, it is copied to the text area to form history of executed commands. In addition, the entire list of commands in the text area can be executed by selecting Script->Run menu option. The text area can also be used to paste existing scripts or display imported scripts. |
Sirius command reference
| Command | Description |
| addselect | Adds specified atom set to the existing selection |
| attach | If more than one structure is loaded, it is necessary to specify which structure the commands should apply to. To do so, enter attach <structure name>. Names of loaded structures appear in any appearance dialog's pulldown menu or under File->Close structure selection menu. |
| background | This command followed by color name or comma separated RGB values sets the color of the 3D workspace background. |
| cartoon | Display cartoon ribbon in the current selection. Cartoon ribbon contains flat strands with arrowheads and wide helices. |
| clear | Clears the command panel's text area and command line. |
| close | Closes any loaded structures. |
| color | Sets color of the currently selected atoms. Can be followed by color name (red, blue, etc.), name of the color set (amino, cpk, structure, etc.) or comma-separated RGB triplet enclosed in square brackets. For specific information about predefined color schemes, refer to the RasMol manual. Sample command: color [100,100,230] |
| define | Defines a named set of atoms. The command needs to be followed by name for the new set and the list of atoms or residues that should be in the set. For more complex set definitions (e.g., based on distance), it's recommended to use the interactive approach: Structure->Define/edit atom set. |
| dots | Creates a dot surface around selected atoms. Dot density is defined by the number that follows the keyword. Value of less than 200 produces a medium quality surface, values between 200 adn 600 generate a high quality surface, while higher numbers result in an ultra quality surface (for comparison, refer to the Surface dialog). |
| load | Loads a structure from a file. Command is followed by file name. If full path is not given, it is assumed that the file is located in the current working directory (refer to Preferences). File type is determined from the extension. |
| move | Same as translate - used to move the structure in the direction of coordinate axes. Is added to simplify typing and reduce spelling errors |
| render | Changes rendering state of atoms and bonds in the selection. Is followed by keywords that specify the style and (optional) quality. These are: lines, stick, ballstick and spacefill. Quality can be indicated by adding best keyword after the style specification. If no quality is requested, the structure is rendered as fast. |
| reset | Resets the view to its original location and orientation. Rendering and other appearance is not changed. If a total reset is needed, the structure can simply be reloaded. |
| ribbon(s) | Creates a visible ribbon for selected atoms. In the case of ribbon off, the ribbon is hidden. |
| rotate | Rotates the structure around the specified axis followed by angle in degrees. Direction of rotation is defined by the sign of the angle value. For example, rotate x 45 will rotate the structure around x axis by 45 degrees, with the closest point moving up. Positive value for the y axis indicates that the closest point will move left, and for z axis positive value results in clockwise rotation. |
| select | This command causes a set of atoms to be selected for further operations. A set of atoms can be given in a variety of different methods.
These sets can be combined using comma-separated lists (2,4,5,6,9), dash-separated ranges of residue numbers (100-320), wild cards (*E indicates all atoms in chain E), parentheses and logical operators (and, or, not). AND operator between two sets returns a union of the sets - only atoms present in both sets form the resulting set. E.g. select ala230 and sidechain highlights only side chain atoms in the alanine 230. OR operator adds the two sets. For example, select ala or tyr will select all alanine and tyrosine residues int he structure. NOT operator will exclude atoms of the second set from the atoms of the first set. For example, select ala230 and not ca will highlight the entire residue except for the CA. A full list of predefined named sets is given below. Command addselect follows exactly the same syntax rules, except it doesn't clear the previous selection. |
| set | Used in combination with strands and a numeric value, sets the following strands representation to a thin backbone trace. E.g., set strands 1. |
| spacefill | Renders selected atoms using spacefilling representation. |
| strands | Creates a ribbon for the selected atoms. If a set strands 1 was entered before, a thin backbone trace is created. |
| trace | Creates a uniform ribbon for selected residues. The ribbon has round cross-section. |
| translate | Moves the structure in the direction of one of the three coordinate axes. For example, translate x 10 will move the structure by 10 angstroms to the right. Positive directions for the axes: x - right, y - up, z - closer to the point of view. |
| wireframe | This command indicates whether the atomic structure should be displayed. For example, wireframe off causes the atoms and bonds to become hidden. Just the wireframe command with no additional parameters causes the structure to appear. This command doesn't affect displayed ribbons. |
| zap | Same as close, and used for compatibility with existing RasMol scripts. |
Predefined named sets
| Name | Content |
| at | Adenosine and thymidine |
| acidic | All acidic amino acids |
| acyclic | All acyclic amino acids |
| aliphatic | All aliphatic amino acids |
| alpha | All alpha carbons |
| amino | All aminoacids |
| aromatic | All aromatic amino acids |
| backbone | Backbone atoms in protein or DNA structures |
| basic | All basic amino acids |
| bonded | All atoms that are bonded to at least one other atom |
| buried | All amino acids with propensity to be buried (not their actual location in the structure) |
| cg | Cytosine and guanosine |
| charged | All charged amino acids |
| cyclic | All cyclic amino acids (both aromatic and non-aromatic) |
| helix |
All amino acids that are in a helix (their actual location) |
| hetero | All hetero atoms in the protein/DNA structure (denoted as HETATM in PDB files) |
| hydrogen | All hydrogen atoms |
| hydrophobic | All hydrophobic amino acids |
| large | All amino acids with large side chains |
| ligand | Any residues that are not amino acids, nucleic acids or solvent |
| medium | All amino acids with medium-sized side chains |
| neutral | All neutral amino acids |
| nucleic | All nucleic acids |
| polar | All polar amino acids |
| protein | All amino acids |
| purine | Adenosine and guanosine |
| pyrimidine | Cytosine and thymidine |
| sheet | All amino acids in beta-strands |
| sidechain | Side chains of all amino acids |
| small | All amino acids with small side chains |
| solvent | Solvent atoms |
| surface | Amino acids with propensity to form protein surface (not their actual location) |
| turn | All amino acids in secondary structure turns |
| water | All water molecules |
Predefined color schemes
| Name | Color |
| amino | Color scheme used for amino acid coloring in RasMol |
| structure | Colors residues according to the secondary structure element they are located in. Specific colors that correspond to helices, strands, etc. can be set in Structure Viewer tab of the Preferences dialog. |
| cpk | CPK coloring scheme used in RasMol |